第一步:数据预处理
在本教程中,我们将以SNARE-seq数据 (Chen, et al. 2019) 为例,展示如何为GLUE模型训练准备必要的数据。SNARE-seq数据由成对的scRNA-seq和scATAC-seq图谱组成,但我们将它们视作非配对的,并尝试使用GLUE来对齐这两个组学层。
[1]:
import anndata as ad
import networkx as nx
import scanpy as sc
import scglue
from matplotlib import rcParams
[2]:
scglue.plot.set_publication_params()
rcParams["figure.figsize"] = (4, 4)
读取数据
首先,我们需要把scRNA-seq和scATAC-seq数据准备成 AnnData 对象。 AnnData 是我们在 scglue 中使用的标准数据类。如果您不熟悉如何从头开始构建 AnnData 对象,以及如何将其他格式的数据(csv、mtx、loom等)读入 AnnData 对象,请查看他们的 文档。
这里我们加载现有的 h5ad 文件,这是 AnnData 的本地文件格式。本教程使用的 h5ad 文件可以从这里下载:
http://download.gao-lab.org/GLUE/tutorial/Chen-2019-RNA.h5ad
http://download.gao-lab.org/GLUE/tutorial/Chen-2019-ATAC.h5ad
[3]:
rna = ad.read_h5ad("Chen-2019-RNA.h5ad")
rna
[3]:
AnnData object with n_obs × n_vars = 9190 × 28930
obs: 'domain', 'cell_type'
[4]:
atac = ad.read_h5ad("Chen-2019-ATAC.h5ad")
atac
[4]:
AnnData object with n_obs × n_vars = 9190 × 241757
obs: 'domain', 'cell_type'
预处理scRNA-seq数据
(预计时间:约2分钟)
首先,scRNA-seq表达矩阵应该包含原始的UMI计数:
[5]:
rna.X, rna.X.data
[5]:
(<9190x28930 sparse matrix of type '<class 'numpy.float32'>'
with 8633857 stored elements in Compressed Sparse Row format>,
array([1., 1., 1., ..., 1., 1., 1.], dtype=float32))
在进行预处理之前,我们先将原始UMI计数备份到一个名为“counts”的层中。它将在未来的模型训练中使用到。
[6]:
rna.layers["counts"] = rna.X.copy()
接着按照最基础的 scanpy 流程进行数据预处理(如果您不熟悉,请查看他们的 教程)。
首先,我们使用“seurat_v3”方法选择2000个高变异基因。
[7]:
sc.pp.highly_variable_genes(rna, n_top_genes=2000, flavor="seurat_v3")
然后,对数据进行归一化和缩放,以及PCA降维,默认使用100个主成分。
PCA嵌入将被用于 第二步,作为编码器的第一个转换,以减少模型大小。
[8]:
sc.pp.normalize_total(rna)
sc.pp.log1p(rna)
sc.pp.scale(rna)
sc.tl.pca(rna, n_comps=100, svd_solver="auto")
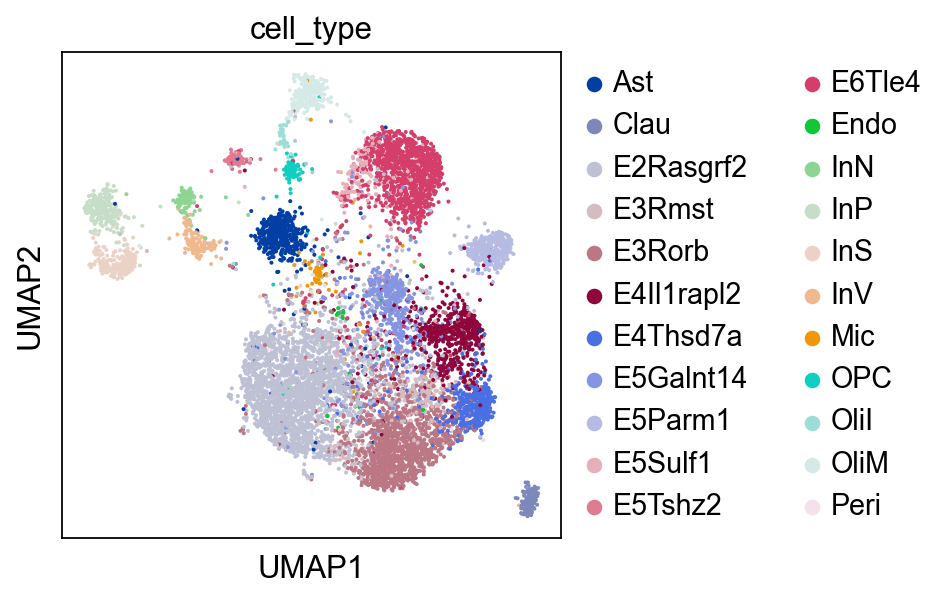
可选的,我们可以用UMAP对RNA模态进行可视化。
[9]:
sc.pp.neighbors(rna, metric="cosine")
sc.tl.umap(rna)
sc.pl.umap(rna, color="cell_type")
预处理scATAC-seq数据
(预计时间:约2分钟)
与scRNA-seq类似,scATAC-seq染色质可及性矩阵也应该包含原始计数。
[10]:
atac.X, atac.X.data
[10]:
(<9190x241757 sparse matrix of type '<class 'numpy.float32'>'
with 22575391 stored elements in Compressed Sparse Row format>,
array([1., 1., 1., ..., 1., 1., 1.], dtype=float32))
对于scATAC-seq,我们使用函数 scglue.data.lsi 进行潜在语义索引(LSI)降维,这是 Signac 中LSI函数的Python重新实现。我们将维度设置为100。另一个关键参数 n_iter=15 被传递给 sklearn.utils.extmath.randomized_svd,将其设置为更大的值可以提高随机SVD的精度。
LSI嵌入将被用于 第二步,作为编码器的第一个转换,以减少模型大小。
[11]:
scglue.data.lsi(atac, n_components=100, n_iter=15)
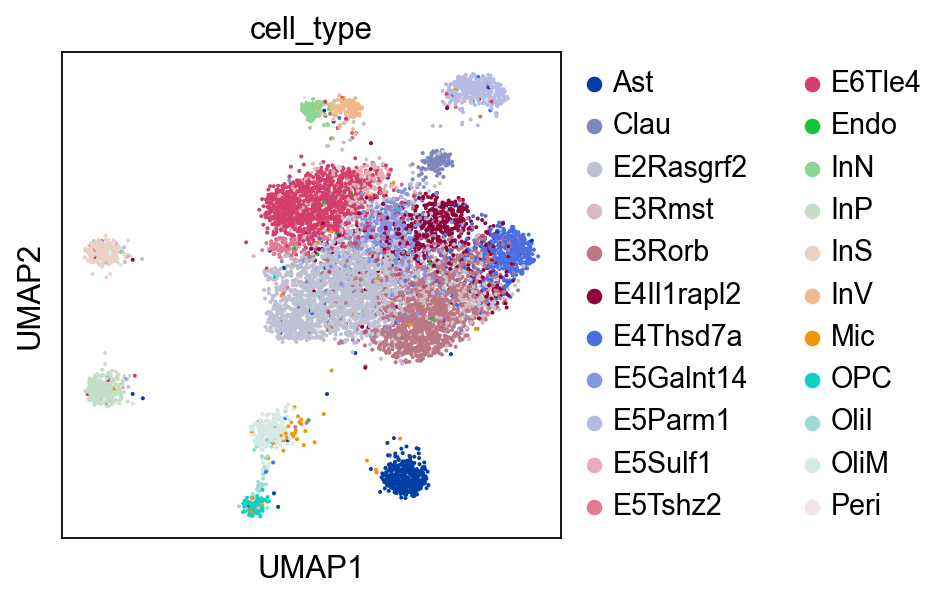
可选的,我们可以用UMAP将ATAC模态可视化。
[12]:
sc.pp.neighbors(atac, use_rep="X_lsi", metric="cosine")
sc.tl.umap(atac)
[13]:
sc.pl.umap(atac, color="cell_type")
构建先验调控图
(预计时间:约2分钟)
接下来,我们需要构建先验引导图,GLUE将利用它来对齐多组学。该图应该包含对应于组学特征(如scRNA-seq的基因和scATAC-seq峰)的节点,以及对应于先验调控关系的边。
GLUE接受 networkx 形式的引导图(如果您不熟悉,请查看 介绍)。因此,原则上您可以根据需要手动构建引导图,只要该图符合以下标准:
图中的边 应该包含“weight”和“sign”作为边的属性,weight的范围是(0,1],sign的值是1或-1。
图应该包含每个节点的自环,其中weight = 1 且 sign = 1。
建议使用无向图 (Graph, MultiGraph),或对称的有向图 (DiGraph, MultiDiGraph)。
下面,我们将展示如何使用 scglue 中的内置函数为scRNA-seq和scATAC-seq整合构建引导图。
获取基因组坐标
关联ATAC峰与基因最常用的先验信息是基因组的上的距离。因此,我们需要ATAC峰和基因的基因组坐标,并将其作为特征元数据存储在 var 中。
对于典型scRNA-seq数据集,只有基因名称或者基因ID被存储,而没有基因组坐标,比如下面这个例子:
[14]:
rna.var.head()
[14]:
| highly_variable | highly_variable_rank | means | variances | variances_norm | mean | std | |
|---|---|---|---|---|---|---|---|
| genes | |||||||
| 0610005C13Rik | False | NaN | 0.001415 | 0.001413 | 0.958918 | 0.000832 | 0.024802 |
| 0610009B22Rik | True | 1528.0 | 0.017301 | 0.022228 | 1.126013 | 0.010283 | 0.091634 |
| 0610009E02Rik | False | NaN | 0.014799 | 0.017193 | 1.023411 | 0.008182 | 0.076223 |
| 0610009L18Rik | False | NaN | 0.016540 | 0.020403 | 1.082735 | 0.009849 | 0.088678 |
| 0610010F05Rik | False | NaN | 0.157563 | 0.197176 | 1.004244 | 0.086493 | 0.245927 |
所以,我们提供了一个实用函数 scglue.data.get_gene_annotation 来补充GTF文件中的坐标信息。下面的例子假设 rna.var_names 与GTF文件中的“gene_name”属性相对应,对于其他情况,请查看 函数文档。
这里使用的GTF文件可以从 GENCODE 下载。
[15]:
scglue.data.get_gene_annotation(
rna, gtf="gencode.vM25.chr_patch_hapl_scaff.annotation.gtf.gz",
gtf_by="gene_name"
)
rna.var.loc[:, ["chrom", "chromStart", "chromEnd"]].head()
[15]:
| chrom | chromStart | chromEnd | |
|---|---|---|---|
| genes | |||
| 0610005C13Rik | chr7 | 45567793 | 45575327 |
| 0610009B22Rik | chr11 | 51685385 | 51688874 |
| 0610009E02Rik | chr2 | 26445695 | 26459390 |
| 0610009L18Rik | chr11 | 120348677 | 120351190 |
| 0610010F05Rik | chr11 | 23564960 | 23633639 |
注意,坐标列名是“chrom”,“chromStart”和“chromEnd”,与 BED格式 前三列相对应。这些精确的列名是必须的,像“chr”,“start”,“end”这样的替代名称**不被识别**。
对于scATAC-seq数据,坐标已经在 var_names 中,我们只需要提取它们。
[16]:
atac.var_names[:5]
[16]:
Index(['chr1:3005833-3005982', 'chr1:3094772-3095489', 'chr1:3119556-3120739',
'chr1:3121334-3121696', 'chr1:3134637-3135032'],
dtype='object', name='peaks')
[17]:
split = atac.var_names.str.split(r"[:-]")
atac.var["chrom"] = split.map(lambda x: x[0])
atac.var["chromStart"] = split.map(lambda x: x[1]).astype(int)
atac.var["chromEnd"] = split.map(lambda x: x[2]).astype(int)
atac.var.head()
[17]:
| chrom | chromStart | chromEnd | |
|---|---|---|---|
| peaks | |||
| chr1:3005833-3005982 | chr1 | 3005833 | 3005982 |
| chr1:3094772-3095489 | chr1 | 3094772 | 3095489 |
| chr1:3119556-3120739 | chr1 | 3119556 | 3120739 |
| chr1:3121334-3121696 | chr1 | 3121334 | 3121696 |
| chr1:3134637-3135032 | chr1 | 3134637 | 3135032 |
图构建
现在我们有了所有组学层特征的基因组坐标,可以使用 scglue.genomics.rna_anchored_guidance_graph 函数来构建引导图。
默认情况下,如果ATAC峰与基因体或启动子区域重叠,就会与该基因相连。关于可以调整的设置,请查看 函数文档。
[18]:
guidance = scglue.genomics.rna_anchored_guidance_graph(rna, atac)
guidance
[18]:
<networkx.classes.multidigraph.MultiDiGraph at 0x7f8e003f1610>
下面通过 scglue.graph.check_graph 函数验证所获得的引导图符合之前所有的先验图构建标准。
[19]:
scglue.graph.check_graph(guidance, [rna, atac])
[INFO] check_graph: Checking variable coverage...
[INFO] check_graph: Checking edge attributes...
[INFO] check_graph: Checking self-loops...
[INFO] check_graph: Checking graph symmetry...
[INFO] check_graph: All checks passed!
同时,通过标记引导图中与高变异基因相连通的ATAC峰,可以将高变异特征传播到ATAC模态:
[20]:
atac.var.head()
[20]:
| chrom | chromStart | chromEnd | highly_variable | |
|---|---|---|---|---|
| peaks | ||||
| chr1:3005833-3005982 | chr1 | 3005833 | 3005982 | False |
| chr1:3094772-3095489 | chr1 | 3094772 | 3095489 | False |
| chr1:3119556-3120739 | chr1 | 3119556 | 3120739 | False |
| chr1:3121334-3121696 | chr1 | 3121334 | 3121696 | False |
| chr1:3134637-3135032 | chr1 | 3134637 | 3135032 | False |
如果 rna_anchored_guidance_graph 函数不能满足需要(例如想加入实验鉴定的调控证据),您可能需要手动构建引导图。我们也提供了一些更底层的实用工具,比如 scglue.genomics.window_graph, scglue.graph.compose_multigraph, scglue.graph.reachable_vertices。您可以参考我们的 案例研究,其中我们将基因组线性距离与pcHi-C和eQTL证据相结合,构建了一个混合先验调控图。
保存预处理过的数据文件
最后,我们保存预处理过的数据,供 第二步 使用。
[21]:
rna.write("rna-pp.h5ad", compression="gzip")
atac.write("atac-pp.h5ad", compression="gzip")
nx.write_graphml(guidance, "guidance.graphml.gz")